
What causes ASXL-related disorders?
Article summary
ASXL-related disorders, which include Bohring-Opitz Syndrome (ASXL1), Shashi-Pena Syndrome (ASXL2), and Bainbridge-Ropers Syndrome (ASXL3), occur because of differences or variants in a person’s DNA and genes. Variants in a gene can lead to problems with protein production which can result in symptoms of disease. Variants in ASXL genes change the way that many other genes are regulated. Because the genes regulated by ASXL play key roles in the function of multiple organ systems, such as in the heart, brain, lungs, and kidneys, many parts of the body can be affected by ASXL variants.
The mutations in ASXL genes usually happen randomly during the process of cell replication and are not inherited from a parent – this is known as a de novo variant. Rarely, ASXL-related disorders can be inherited through gonadal mosaicism (also called germline mosaicism), which is when a person has two or more genetically distinct cell lines in their eggs or sperm cells. If a person with an ASXL variant were to have children, there is a 50% chance that their offspring would inherit the same ASXL variant and have the same condition as the parent. (This is known as an autosomal dominant inheritance pattern.) The type of pathogenic (disease-causing) variant in ASXL-related disorders is called a truncating variant. Truncating variants result in shortened, non-functional proteins that have serious functional consequences leading to the symptoms seen in ASXL-related disorders.
Genetics 101 and ASXL-related disorders
ASXL-related disorders occur because of differences in a person’s DNA. But what exactly does DNA do? And why do genes matter? The answers lie in the trillions of cells that make up the human body.
Each cell in our body contains chromosomes. Most people have 46 chromosomes, with half coming from the egg and half from the sperm. Chromosomes are made up of tightly wound DNA, which provides specific instructions to the cells for how to make proteins. DNA is segmented into genes. Each gene then has its own specific functions and gives instructions to, or codes, for the production of certain proteins.
These proteins, which are created by the instructions they get from genes, are essential for the normal functioning of the human body. Sometimes, when there is a change in a gene, the corresponding protein is not made correctly. You may hear or see these gene changes referred to as variants or mutations. Variants in a gene can lead to not enough protein being made, misshapen proteins, incorrect proteins, or even too much protein. Differences in proteins can lead to human disease. Disease-causing variants are called pathogenic variants. People with an ASXL-related condition typically have one normal copy of the ASXL gene and one copy that contains a mutation in the ASXL gene.
The ASXL genes, specifically ASXL1 (Bohring-Opitz Syndrome), ASXL2 (Shashi-Pena Syndrome), and ASXL3 (Bainbridge-Ropers Syndrome), play a role in the genetic regulatory system. ASXL genes code for proteins that help regulate our DNA. While all people have ASXL genes, there are a small number of people born with variants in these genes. Variants in ASXL genes change the way that many other genes are regulated. You can think of the ASXL genes like a quarterback on a football team. The quarterback is necessary to distribute the ball to other players on the field; however, if the quarterback suffers an injury, the other players may not receive the ball and the team may struggle.
Because other genes regulated by ASXL play key roles in the function of multiple organ systems, such as in the heart, brain, lungs, and kidneys, many parts of the body can be affected by ASXL variants. Therefore, patients with Bohring-Opitz, Shashi-Pena, and Bainbridge-Ropers Syndromes experience symptoms in many different organs.
Read more: Epigenetics and ASXL-related disorders

Each cell contains chromosomes. Chromosomes are made up of tightly wound DNA. DNA is segmented into genes.
Image credit: https://nci-media.cancer.gov/pdq/media/images/793768.jpg
How do genetic differences arise in ASXL-related disorders?
The mutations in ASXL genes usually happens randomly during the process of cell replication shortly after an egg and sperm cell meet and are not usually inherited from a parent. When a gene variant arises spontaneously and is not passed down from either parent, the variant is characterized as de novo. De novo in Latin literally means “anew” and “from the beginning”.
Some parents may still wonder if they played a role in causing the de novo variant. The truth is that there is no way to prevent or cause an ASXL de novo variant – the variants arise completely randomly and are outside of our control.
Rarely, ASXL-related disorders can be inherited through gonadal mosaicism (also called germline mosaicism). Gonadal mosaicism is the phenomenon in which a person has two or more genetically distinct cell lines in their eggs or sperm cells. In other words, a person could have a genetic change in some but not all of their cells, and therefore have none or only limited symptoms of the genetic condition. If that person contributes an egg or sperm cell with the genetic change to a fetus, the offspring may have symptoms of a genetic condition that the contributing parent does not have.
Mosaicism is difficult to test for in parents since each sperm or egg cell could have varying degrees of mosaicism (and it is not currently possible to look at each individual sperm or egg a person has). Because the possibility of germline mosaicism exists, the general risk for parents with one child affected by an ASXL-related disorder to have another affected child is 1%. However, there have been several reported instances of familial recurrence. The current estimated risk of recurrence is 1-5%.
Can ASXL-related disorders be passed down?
If a person with an ASXL variant were to have children, there is a 50% chance that their offspring would inherit the same ASXL variant and have the same condition as the parent. This is known as an autosomal dominant inheritance pattern.
In autosomal dominant inheritance, a person only needs one copy of the gene variant to have the condition. For example, a person with Bohring-Opitz syndrome has one chromosome that contains an ASXL1 variant and another chromosome that is completely normal. (Recall that we have 23 pairs of chromosomes with 23 chromosomes coming from the egg and 23 chromosomes coming from the sperm.) If this person were to reproduce, they would pass down either chromosome to their offspring. Whether they pass down the chromosome with a variant or the normal chromosome is essentially a coin flip. If the child inherited the chromosome with the ASXL variant, they would also have the condition. If the child inherited the normal chromosome, they would not have the condition.
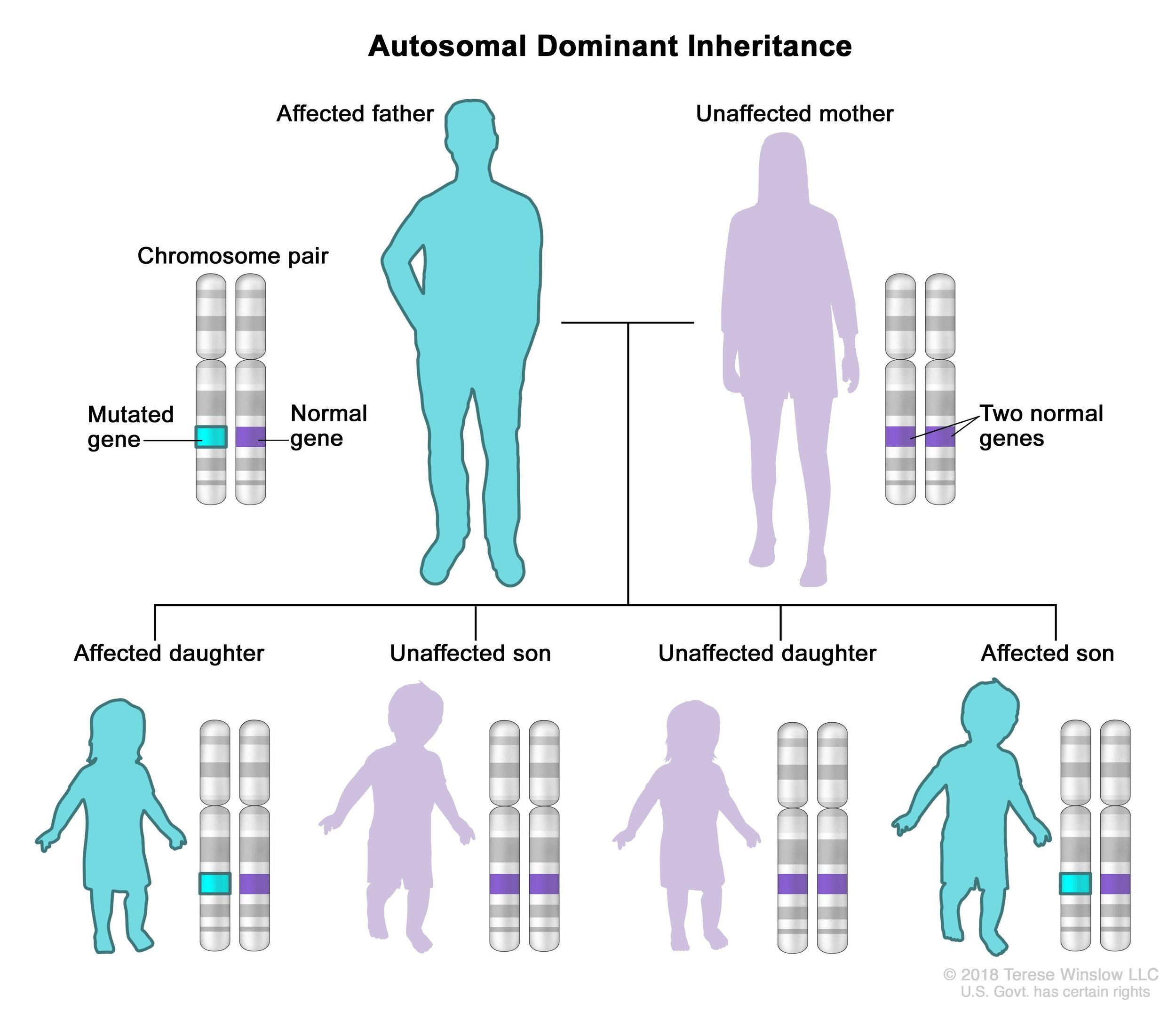
An illustration of the autosomal dominant inheritance pattern where an affected parent has a 50% chance of passing the disorder to a child
Image credit: https://www.nia.nih.gov/health/alzheimers-disease-genetics-fact-sheet
Types of ASXL variants
Gene variants are not all the same. In fact, the type of variant determines the way a protein is made. (Recall, genes provide the instructions for how proteins are made, and proteins are essential for the function of the human body.) Gene variants can cause proteins to be made incorrectly, made in excess, or not made at all.
We believe the type of gene variant that causes ASXL-related disorders is called a truncating loss-of-function variant. In truncating variants, the DNA sequence is stopped prematurely which leads to a shorter, nonfunctional protein which leads to the serious functional consequences that cause the symptoms seen in ASXL-related disorders.
Acknowledgements

Thank you to Elian Silverman, a genetic counseling student, who wrote this article. We are are grateful to Debbie Requesens and the JumpStart Program of the Orphan Disease Center for pairing Elian with us for this project.
We also extend our gratitude to Dr. Wen-Hann Tan and Dr. Bianca Russell for providing medical review of this article.